



















A Doença de Behçet é uma vasculite sistêmica idiopática de pequenos vasos, que pode envolver artérias e veias de todos os calibres. O nome é em homenagem ao dermatologista turco Hulusi Behçet – quem primeiro descreveu a doença em 1937.
Classicamente se apresenta com a tríade: úlceras aftosas + úlceras genitais + uveíte. Veja mais sobre os sinais e sintomas, e como tratar pacientes com a doença.
O que é a Doença de Behçet?
A Doença de Behçet é uma vasculite sistêmica idiopática rara, que afeta cerca de 5 pessoas para cada milhão por ano. Tem pico de incidência entre 25-35 anos e afeta igualmente ambos os sexos.
Vamos lembrar: vasculites sistêmicas são doenças em que há inflamação da parede de vasos sanguíneos, levando a sinais/sintomas locais e constitucionais. São secundárias quando decorrem de outras doenças (como lúpus) ou primárias quando a causa é desconhecida (idiopática) – sendo a doença a manifestação por si só.
Embora de causa desconhecida, sabe-se que fatores genéticos estão envolvidos na doença. Os principais alelos relacionados são do HLA, sendo o principal exemplo o HLA-B51. Porém, apenas o fator genético não é capaz de desenvolver a doença. São necessários gatilhos para o aparecimento de Behçet em indivíduos geneticamente suscetíveis – levando a uma resposta inflamatória autoperpetuante.
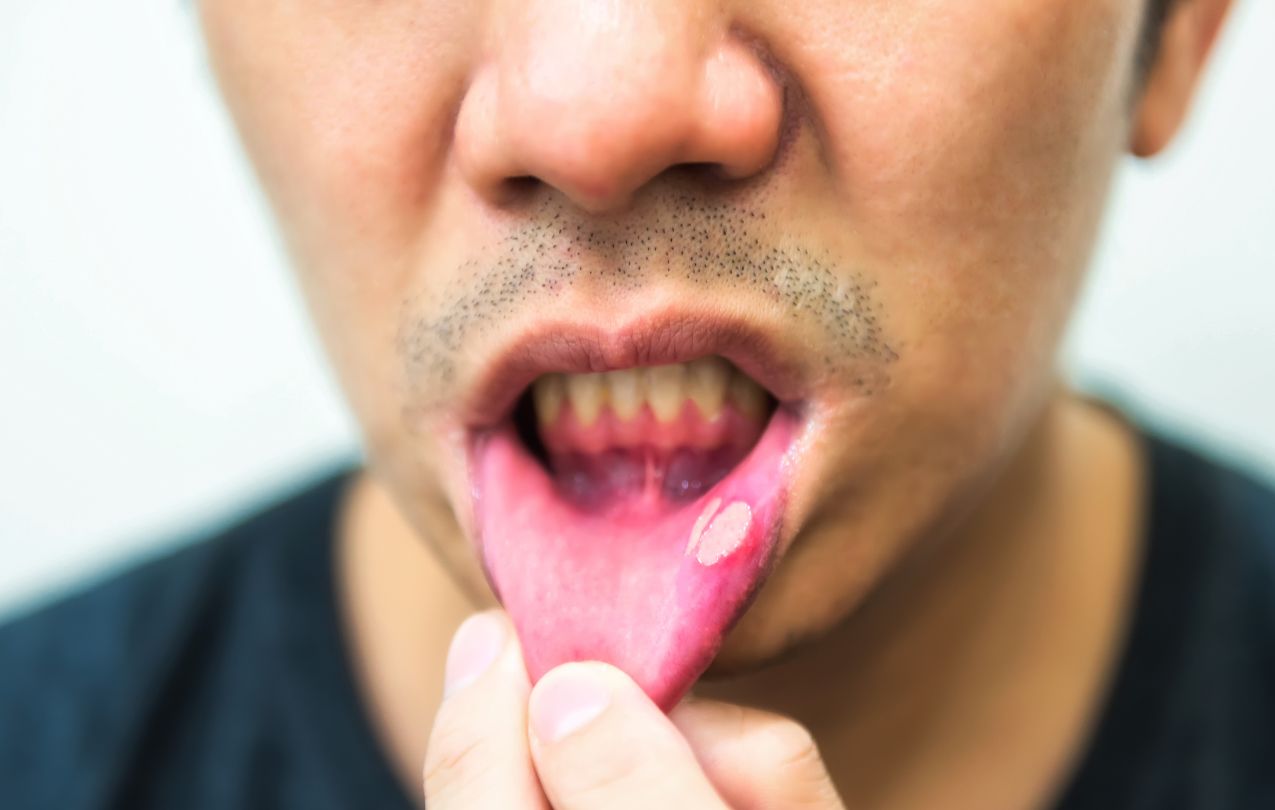
Quais os sinais e sintomas?
O quadro clínico é tipicamente marcado por períodos de melhora e de piora. Como citamos, achados específicos incluem úlceras aftosas + úlceras genitais + uveíte. Sintomas inespecíficos incluem: fadiga, febre, perda de peso, dores articulares, musculares etc.
As úlceras aftosas orais são tipicamente o primeiro achado, e o mais comum. As aftas são recorrentes, dolorosas, com diâmetro de 2-10 mm, apresentando base amarelada-necrótica, circunscrita por halo eritematoso. Podem ser únicas ou múltiplas, acometendo mucosa orofaríngea. Não há formação de cicatrizes, desaparecendo entre 1-3 semanas.
O aparecimento das úlceras genitais é menos habitual. São semelhantes às úlceras aftosas orais, porém não são dolorosas e tipicamente deixam cicatrizes. O acometimento típico é na região da bolsa escrota, pênis, vulva, vagina, colo uterino e ainda região perianal – poupando glande e uretra.
Lembrar que apesar das úlceras aftosas serem mais comuns na região da orofaringe e genitais, elas também podem acometer qualquer região do trato gastrointestinal.
Há também lesões cutâneas, presentes em quase todos os pacientes e de diversas formas: pseudofoliculite, lesões acneiformes em face, lesões papulopustulosas e eritema nodoso.

O envolvimento ocular tipicamente se inicia após as manifestações mucocutâneas. É progressivo, recorrente, com curso crônico. O achado mais característico é uveíte anterior com hipópio (presença de pus na região anterior do olho).
Dentre os achados vasculares, citamos tromboflebites superficiais, TVP (5%), trombose de veia cava, Síndrome de Budd-Chiari, tromboses cerebrais e aneurismas de artéria pulmonar.
Em até 10% dos pacientes pode haver envolvimento do sistema nervoso central. São vários os possíveis achados: meningite asséptica, síndrome piramidal, ataxia cerebelar, acidente isquêmico transitório etc.
Leia também: Como diagnosticar a Doença de Behçet: critérios ISG de 1990.
Como tratar a Doença de Behçet?
Muitas das propostas terapêuticas não possuem um embasamento científico consolidado, sendo suportado empiricamente por opiniões de especialistas. As apresentações clínicas da doença podem ser as mais variadas possíveis, de casos leves a muito graves – sendo o tratamento variável em decorrente disso.
Acometimento osteoarticular
Na presença de acometimento osteoarticular, como artrite, opta-se inicialmente pela escolha de um AINE (anti-inflamatório não esteroidal) ou colchicina. Na refratariedade, opta-se pelo uso de corticoides sistêmicos em doses baixas.

Manifestações ulcerosas
Para as manifestações ulcerosas (aftas orais/genitais) iniciar-se a abordagem com glicocorticoides tópicos nas lesões (p.ex. Triancinolona tópica 3x ao dia). Comumente não há resposta satisfatória com agentes tópicos isoladamente, por isso, opta-se pela associação com colchicina.
Se não houver melhora com a associação de corticoide tópico e a colchicina, ou caso o paciente apresente múltiplas lesões, opta-se pela prescrição de corticoide sistêmico.
Eventualmente alguns pacientes vão necessitar de uso de baixas doses de corticoide por longos períodos. No entanto, quando a dose necessária de manutenção for maior que 10 mg/dia, opta-se pela associação de doses menores de corticoide (que devem ser retirados quando possível) com outros imunomoduladores.
Um medicamento muito promissor para o controle de úlceras é o apremilaste (OTEZLA®) – é um inibidor da fosfodiesterase-4, tendo ação imunomodulatória. Necessita, porém, de mais estudos para avaliar sua real eficácia.
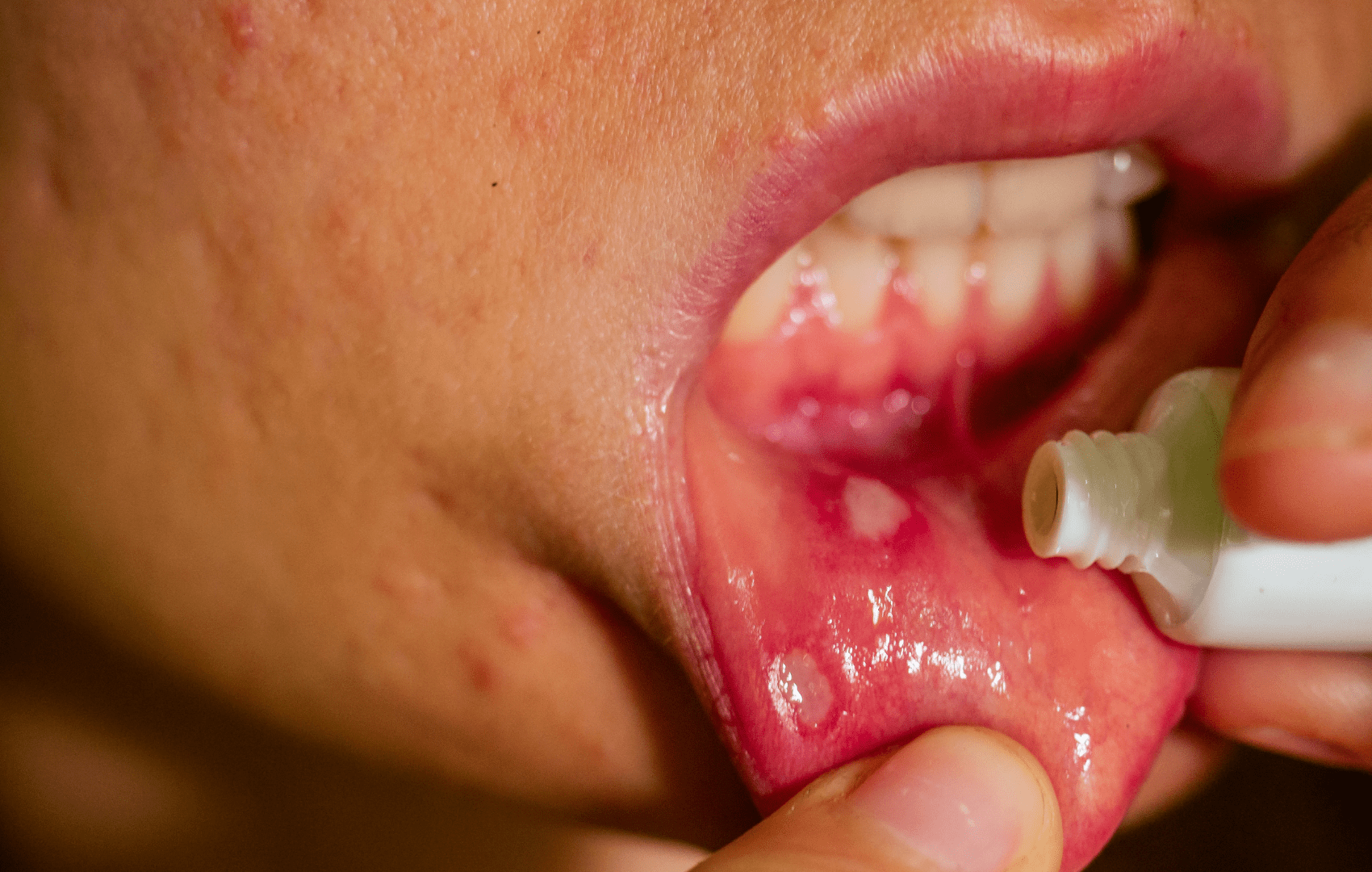
Manifestações dermatológicas
De maneira geral, para os achados dermatológicos, iniciamos com colchicina. Na presença de lesões moderadas-graves ou refratárias, associa-se um corticoide sistêmico.
No caso de lesões de eritema nodoso, opta-se pela associação de prednisona com azatioprina. O corticoide deve ser retirado gradualmente assim que possível.
Manifestações oftalmológicas
As lesões oculares são tratadas de acordo com a gravidade. As duas manifestações mais comuns são a uveíte anterior e a posterior, tendo tratamentos diferentes.
A uveíte anterior é geralmente tratada com glicocorticoides tópicos associados com colírio midriático/ciclopégico. Pacientes que não apresentam melhora, deverão receber corticoides sistêmicos.
A uveíte posterior é uma séria ameaça a perda de visão e requer uma intensiva imunossupressão. Para isso, utilizamos corticoide em doses associado ao imunomodulador azatioprina. Paciente sem uma resposta adequada, ou aqueles com casos graves, adicionamos um inibidor da TNF-alfa.
A uveíte anterior isolada é rara, sendo o mais usual a presença de panuveíte (anterior + intermediária + posterior) – com o tratamento sendo uma combinação de ambas. O tratamento com os imunomoduladores/imunossupressores devem continuar por pelo menos 18-24 meses, dependendo da resposta obtida.

Manifestações neurológicas
Alterações neurológicas são potencialmente graves e requerem uma intensiva imunossupressão. Para isso, o esquema é o mesmo da uveíte posterior: utilizamos corticoide em doses associado a azatioprina e, na ausência de resposta adequada ou casos graves, adiciona-se um inibidor da TNF-alfa ao esquema.
Outras manifestações
Para tratamento de nefrite mínima ou leve, não há necessidade de um tratamento específico. Porém, deve-se manter seguimento para avaliar possíveis agravamentos. Em casos moderados ou graves, o tratamento sugerido é com colchicina.
Embora bastante raro, na Doença de Behçet pode haver dilatações aneurismáticas arteriais de médio e grande calibre. O tratamento inicial envolve a prescrição de um corticoide sistêmico associado a um imunomodulador, tipicamente a ciclofosfamida. Em acometimentos graves, procedimentos invasivos como angioplastia transluminal percutânea e/ou bypass cirúrgicos podem ser necessários.
Na presença de tromboses, essas devem ser tratadas da mesma forma que se trata nos demais indivíduos – não tendo, portanto, diferença terapêutica.
Quer saber mais sobre a Doença de Behçet, incluindo fisiopatologia, diagnóstico e exames complementares? Acesse nosso app WeMEDS®. Disponível na versão web ou para download para iOS ou Android.
—
Referências:
International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. 1990; 335: 1078-80.
Smith, EL. – Clinical manifestations and diagnosis of Behçet’s syndrome. In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA.
Smith, EL. – Treatment of Behçet’s syndrome. In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA.
















{kind=link}
[…] como a prednisona, são comuns no tratamento inicial9. Eles podem ser usados de várias formas, dependendo da necessidade9. A […]