



















A Síndrome DiGeorge também pode ser chamada de Síndrome de Deleção do cromossomo 22q11.2 ou ainda Síndrome Velocardiofacial. É uma doença cromossômica em que há alterações em diversos genes, portanto, seus efeitos são variados.
Os pacientes podem apresentar uma ampla gama de fenótipos clínicos, o que pode dificultar o diagnóstico. Entretanto, um dos seus principais efeitos é a alteração no timo, o que prejudica o sistema imune da criança devido a uma redução dos linfócitos T.
Quais as causas da Síndrome DiGeorge?
A síndrome DiGeorge é uma condição genética em que ocorre uma deleção em uma região do cromossomo 22 (22q11.2). Essa deleção pode ser de vários tamanhos, o que se reflete nos fenótipos variáveis. A maioria dos pacientes (cerca de 90%) mostra uma deleção que envolve aproximadamente 40 genes.
Os genes causadores de doenças mapeados para a região abrangida estão envolvidos em processos do ciclo celular, do metabolismo celular e da metilação do DNA, entre outras funções. Muitos dos genes afetados possuem funções regulatórias no genoma, o que torna o fenótipo ainda mais complexo.
Essa deleção cromossômica altera o desenvolvimento das estruturas que se originam das terceira e quarta bolsas faríngeas no estágio germinal. Essas bolsas faríngeas são um precursor embrionário comum para o timo, as glândulas paratireoides e as regiões conotruncais do coração. Ou seja, esses órgãos são afetados na síndrome em questão.

Cuidados na pediatria
Com uma frequência média estimada em 1:4000 nascimentos vivos, a síndrome 22q11.2 tem um impacto global e notável nos pacientes afetados, em suas famílias, na sociedade e nos prestadores de cuidados de saúde.
Vale ressaltar que a síndrome 22q11.2 é uma importante causa de morbidade e mortalidade ao longo da vida, e tanto defeitos congênitos importantes quanto complicações que se desenvolvem mais tarde na infância ou até mesmo na idade adulta tornam um diagnóstico definitivo precoce de importância primordial.
Além de ser a causa mais comum de anomalias palatais sindrômicas e disfunção velofaríngea, a síndrome 22q11.2 é também a causa mais comum de esquizofrenia e a segunda causa mais comum de atraso no desenvolvimento e doença cardíaca congênita após a Síndrome de Down, bem como uma causa frequente de doença cardíaca conotruncal.

Sinais e sintomas
Várias desordens clínicas compõem a síndrome 22q11.2, como a síndrome velocardiofacial (disfunção faríngea, anomalia cardíaca, dismorfismo facial), a síndrome de DiGeorge (anomalia cardíaca, hipoparatireoidismo, hipoplasia tímica), e um grupo de desordens descritas pelo acrônimo CATCH22 (defeito cardíaco, características faciais anormais, hipoplasia tímica, fissura palatina, hipocalcemia). A nomenclatura pode ser confusa devido à notável heterogeneidade do fenótipo clínico e variabilidade intersubjetiva da síndrome.
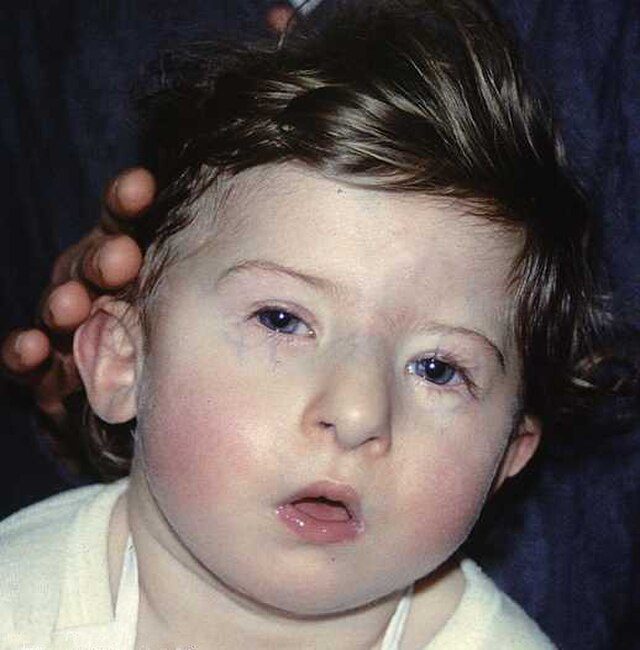
O dismorfismo craniofacial na síndrome 22q11.2 é caracterizado por uma multiplicidade de características fenotípicas faciais. Algumas dessas características faciais não são facilmente reconhecidas clinicamente em crianças, pois são leves por natureza, e a idade é um determinante importante da morfologia facial.
Essas características incluem um rosto alongado, hipertelorismo, ponte nasal larga, pálpebras com aparência de capuz, fendas palpebrais inclinadas para cima, epicanto, nariz longo com ponta bulbosa, base alar estreita, filtro curto, boca pequena, micrognatia e orelhas pequenas, baixas e rotacionadas para trás.
A postura retrognática da mandíbula, o deslocamento interno da parte inferior do rosto e da região ocular, e um ângulo da base craniana aumentado foram identificados como as características de dismorfologia mais importantes na análise cefalométrica e na análise tridimensional densa espacial.
Entre os distúrbios do desenvolvimento orofaríngeo, a palatosquise é uma característica fenotípica frequentemente observável, ocorrendo na forma de fenda aberta, fenda submucosa, desproporção palatofaríngea e língua fendida.
Os distúrbios do desenvolvimento das vias aéreas superiores na síndrome 22q11.2 incluem anomalias laríngeas congênitas. A atresia laríngea devido a uma membrana glótica ou estenose laríngea parcial, a laringomalácia, a fenda laríngea e as anormalidades nas pregas vocais são partes características das características sindrômicas.
A cardiopatia congênita foi reconhecida em aproximadamente 60-80% das crianças afetadas com a síndrome 22q11.2. O subconjunto mais comumente ocorrente de anomalia cardíaca é um defeito conotruncal, como tetralogia de Fallot, atresia pulmonar com comunicação interventricular, tronco arterial comum, arco aórtico interrompido tipo B, defeitos septais conoventricular e/ou atrial, e anomalias do arco aórtico.
As complicações cardiovasculares são a causa mais comum de morte prematura em adultos com a síndrome 22q11.2. Condições associadas importantes, como hipocalcemia, distúrbios da tireoide, doenças autoimunes, problemas comportamentais e deficiência de neurodesenvolvimento, contribuem para o pior desfecho.

Alterações no sistema imunológico
A imunodeficiência é uma característica-chave da síndrome 22q11.2, sendo secundária à aplasia ou hipoplasia do timo, que prejudica o desenvolvimento dos timócitos.
Na síndrome 22q11.2, o subdesenvolvimento do timo ocorre devido à migração prejudicada das células da crista neural para o ectoderma da bolsa. Como ocorre uma migração tímica anormal, os pacientes apresentam reduções leves a moderadas no número de células T.
É visto um amplo espectro de alterações nas células T na síndrome 22q11.2, variando de quase normal a quase completamente imunodeficiente. Também há alteração nos linfócitos B, com os pacientes apresentando baixa produção de imunoglobulinas.
Desordens autoimunes foram descritas em até 23% dos pacientes pediátricos com a síndrome, manifestando-se como doença tireoidiana autoimune, artrite idiopática juvenil, citopenia autoimune (trombocitopenia, anemia hemolítica, neutropenia), doença celíaca, psoríase, vitiligo, hepatite autoimune e doença inflamatória intestinal.
Também foi visto que transtornos psicóticos, regressão do desenvolvimento e comprometimento cognitivo em crianças com síndrome 22q11.2 podem ter uma relação causal com encefalite autoimune.
A disfunção do timo e as anormalidades imunofenotípicas nos compartimentos de células T na síndrome 22q11.2 tornam as crianças afetadas suscetíveis a vírus cancerígenos, como o vírus Epstein-Barr (EBV) e o papilomavírus humano (HPV), que podem estar ligados a um risco aumentado de transformação maligna.

Diagnóstico
A síndrome 22q11.2 é uma síndrome altamente variável, com fenótipos diferentemente expressos, com variabilidade interfamiliar e intrafamiliar ampla em pacientes que compartilham as mesmas bases genéticas.
Isso se deve tanto à notável complexidade da região 22q11.2, bem como aos fatores epigenômicos e ambientais que influenciam a variabilidade fenotípica. Todos esses fatores genéticos, juntamente com a sintomatologia em desenvolvimento relacionada à idade, contribuem para os desafios diagnósticos em pacientes pediátricos com a síndrome 22q11.2.
O diagnóstico da síndrome 22q11.2 tem sido tradicionalmente baseado no reconhecimento de características clínicas e testes citogenéticos usando a técnica de hibridização in situ fluorescente (FISH).

Entretanto, apesar de a FISH ser percebida como o método de teste genético padrão-ouro para confirmar o diagnóstico de síndromes de microdeleção, ela apresenta algumas questões problemáticas: baixa precisão clínica, a baixa taxa confirmatória e a falha em detectar outras microdeleções além da microdeleção alvo são as principais desvantagens deste método. Admite-se que a FISH é barata, mas ainda assim um procedimento altamente trabalhoso e demorado.
Outros métodos são utilizados, como hibridização genômica comparativa (CGH), amplificação de sonda dependente de ligação múltipla (MLPA), reação em cadeia da polimerase quantitativa em tempo real multiplex (qPCR) e análise de microarray de polimorfismo de nucleotídeo único (SNP) de alta resolução.
Entre os genes afetados pela microdeleção, encontra-se o gene TBX1. Portanto, em pacientes com doença clinicamente evidente em quem uma deleção da 22q11.2 não foi identificada por testes FISH ou microarray, é recomendado o teste do gene TBX1.

É possível realizar o diagnóstico já no pré-natal?
Embora os fenótipos pós-natais tenham sido amplamente caracterizados e categorizados, o diagnóstico pré-natal de síndrome 22q11.2 permanece desafiador devido a uma baixa taxa de hereditariedade (10% dos casos) e características parentais não reconhecidas.
A imagem ultrassonográfica fetal pode fornecer informações sobre achados característicos para a síndrome 22q11.2, como polidrâmnio, hipoplasia ou aplasia do timo fetal, anomalias do sistema nervoso central, como ventriculomegalia assimétrica e cavum septum pellucidum dilatado, e, raramente, anomalias esqueléticas, entre outras, como talipes bilaterais e vértebras anômalas.
O método padrão-ouro para detectar microdeleções 22q11.2 continua sendo a triagem no primeiro trimestre por microarray cromossômico (CMA), que é realizado em amostras pré-natais obtidas invasivamente, como vilosidades coriônicas e líquido amniótico.
Um avanço técnico importante no rastreamento pré-natal não invasivo para 22q11.2 é o teste de DNA livre de células. São analisados fragmentos de DNA livre de células presentes no plasma materno, que derivam tanto da mãe quanto do embrião como resultado da apoptose do citotrofoblasto. Uma camada externa da placenta é usada para ensaios qualitativos e quantitativos. A fração fetal é triada para trissomia comum e deleção 22q11.2, bem como para outras trissomias e microdeleções raras.
Tecnologias direcionadas, como baseadas em polimorfismo de nucleotídeo único (SNP), análise digital de regiões selecionadas (DANSR) e ensaio de enriquecimento de captura direcionado (TCEA), bem como a metodologia de sequenciamento de alto rendimento em larga escala (MPSS), são técnicas avançadas usadas na análise de DNA livre de células.

Tratamento
A marcada variabilidade fenotípica da síndrome 22q11.2 é acompanhada por uma ampla gama de déficits imunológicos, variando de linfopenia de células T leve a moderada na forma parcial da síndrome de DiGeorge (pDGS) a uma imunodeficiência combinada grave de células T e B na forma completa (cDGS).
Em crianças mais severamente imunocomprometidas, a reconstituição imunológica pode ser alcançada por meio de transplante de timo, fornecendo a capacidade de produzir células T.
Outra abordagem para cDGS é o transplante de células-tronco hematopoiéticas com células T; no entanto, devido à ausência de timo, o enxerto de células T pós-tímicas pode resultar em baixa qualidade de reconstituição imunológica.
Apesar da grande maioria das crianças com a síndrome 22q11.2 ter níveis normais de imunoglobulinas séricas, devido ao grau variável de comprometimento da imunidade celular, elas são suscetíveis a infecções agudas e recorrentes, entre outras, como sinusite, otite média, mastoidite, pneumonia, infecções do trato urinário e infecções virais.
Questões importantes então são levantadas sobre indicações para medidas preventivas contra infecções em crianças que não se qualificam para terapia de reposição de imunoglobulina, mas apresentam linfopenia de células T.
A profilaxia com antibióticos é indicada, incluindo o uso diário ou em dias alternados de amoxicilina ou azitromicina e cotrimoxazol em crianças com linfopenia de células T avançada, representando risco de infecção por Pneumocystis jiroveci.
Considerações finais
O diagnóstico da Síndrome 22q11.2 é complexo, devido à variedade de sintomas, ampla etiologia genética e atuação de fatores ambientais. Dessa maneira, faz-se necessária a conscientização de pediatras e especialistas em diferentes campos da medicina sobre o amplo espectro de características fenotípicas da síndrome 22q11.2 que podem encontrar.
Além disso, deve ser fornecido um cuidado multidisciplinar abrangente aos pacientes, com cardiologistas, cirurgiões cardíacos, endocrinologistas, laringologistas, neurologistas, cirurgiões e geneticistas, sob a supervisão do imunologista clínico.
—
Referências:
Szczawinska-Popłonyk, A. et al. Chromosome 22q11.2 Deletion Syndrome: A Comprehensive Review of Molecular Genetics in the Context of Multidisciplinary Clinical Approach. Int. J. Mol. Sci. 2023, 24, 8317. https://doi.org/10.3390/ijms24098317.





 no câncer de mama ER+: indicações, eficácia e segurança")



. Entenda mais sobre essa classificação.")






{kind=link}