



















A Síndrome de Turner é a anomalia de cromossomos sexuais mais comum em mulheres. Diz respeito a um conjunto de características e condições médicas decorrentes da perda total ou parcial de um cromossomo sexual, resultando na presença de apenas um cromossomo X.
Nesse artigo, vamos revisar os principais conceitos da síndrome, incluindo as diferentes possibilidades genéticas, diagnóstico e prognóstico.
Etiologia e Fisiopatologia
A prevalência estimada da Síndrome de Turner é de 1:2000-3000 nascimentos do sexo feminino, e cerca de 10% de todos os casos de aborto espontâneo apresentam cariótipo condizente com a síndrome. Esses números pode ser maiores, decorrente de pacientes com características leves que podem não ter sido diagnosticadas, ou da ausência de cariotipagem em abortos espontâneos.
A etiologia é genética, com apenas um cromossomo X funcional. Alguns genes dentro do cromossomo X já foram estudados, responsáveis pelas características fenotípicas dos pacientes. A grande maioria das alterações é no braço curto, principalmente em associação com alterações cardiovasculares.
Em cerca de metade das pacientes, há perda total do cromossomo X (45,X) – monossomia do X, enquanto que de 25-50% dos casos são do tipo mosaico. A chance de o paciente nascer e sobreviver são maiores em casos de mosaicismo, e as características clínicas são variáveis.
Ainda, é possível que a Síndrome de Turner ocorra por anomalias genéticas no cromossomo X, reduzindo sua capacidade funcional. Essas alterações podem ou não estar presentes em mosaicismo (algumas células alteradas, outras não).
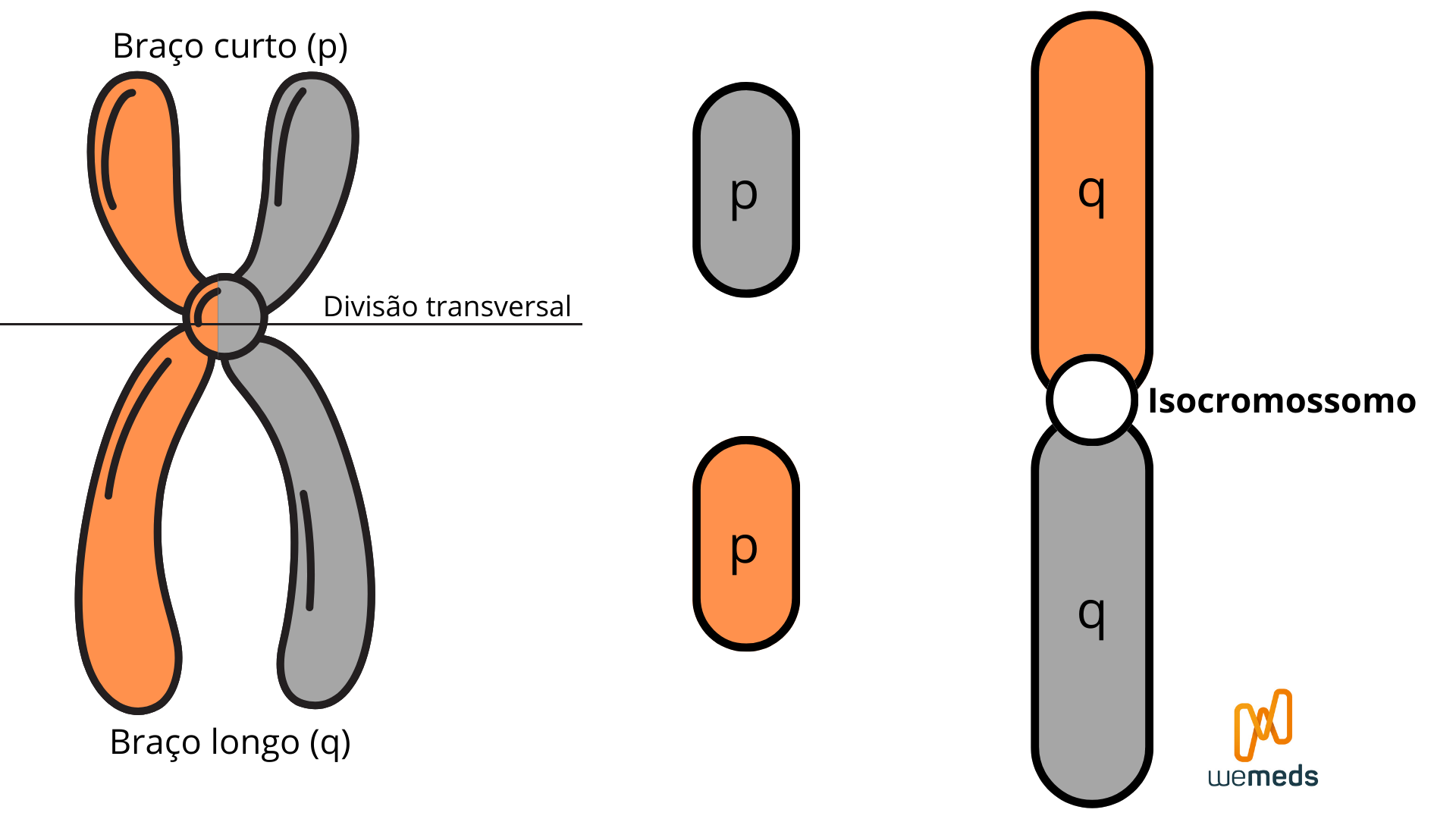
Quando ocorre uma separação transversal das cromátides irmãs, bem no meio do centrômero, há formação de um isocromossomo. Dessa forma, os braços longos cromossômicos se unem formando um cromossomo com 2 cópias de braço longo conectadas. O centrômero fica apenas nesse cromossomo, e os braços curtos são perdidos – chamado de monossomia de braço curto.
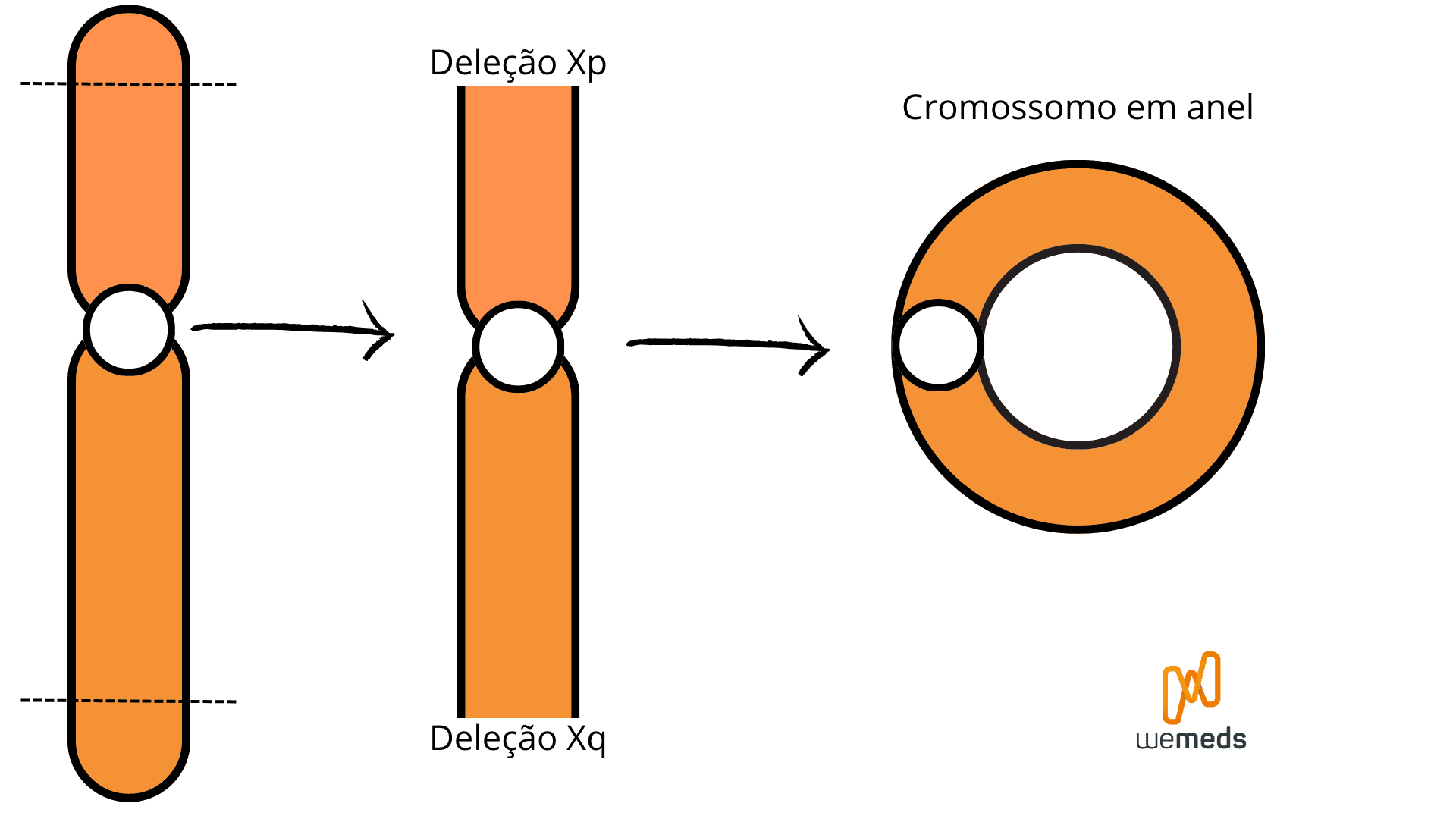
Também pode ocorrer de o cromossomo X perder uma das suas extremidades, seja o braço curto ou o braço longo. Desse caso, estamos diante de uma deleção Xp ou Xq. Quando a deleção ocorre em ambas as extremidades (braço curto E braço longo), as pontas restantes se unem, formando um cromossomo em anel.
Outro mecanismo possivelmente associado à Síndrome de Turner é o imprinting genômico. Nesses casos, mesmo com ambas as cópias do cromossomo X, há expressão preferencial de um deles (materno ou paterno), enquanto o outro está metilado (silenciado).
Independentemente de como ocorre, há um desbalanço gênico pela falta total ou parcial de um cromossomo sexual, resultando apenas em 1 cromossomo X funcional. É uma haploinsuficiência – a presença de apenas um cromossomo não é suficiente para suprir a falta do outro.
Identificando pacientes com Síndrome de Turner
As pacientes com Síndrome de Turner apresentam uma ampla variedade de fenótipos, a depender do tipo de alteração genética. Tipicamente, há baixa estatura, tórax em “escudo” e mamilos distantes, resultando em uma aparência desproporcionalmente larga e atarracada. A insuficiência ovariana é o achado mais comum.
Diferente de grande parte das anomalias genéticas, a inteligência de pacientes com Síndrome de Turner é dentro da média esperada – apenas 10% apresentam atraso de desenvolvimento.
Muitas vezes, o diagnóstico não ocorre no pré-natal ou em neonatos, especialmente em casos leves. Algumas vezes, a doença é identificada em coleta de vilosidade coriônica e/ou amniocentese por outras razões.

Deve-se suspeitar no pré-natal, a partir da 9ª semana, na presença de espessamento da nuca, anomalias renais, fêmur curto, hidropsia fetal, dentre outros achados. Em neonatos, considerar em casos de linfedema de pés e mãos, pescoço alado, displasia ungueal e palato arqueado alto.
Crianças do sexo feminino com alteração inexplicável do crescimento, pescoço alado, linfedema, palato arqueado alto e outras características clássicas da síndrome devem ser investigadas, assim como adolescentes do sexo feminino com baixa estatura, aparência desproporcionalmente larga ou atarracada e imaturidade sexual.
O diagnóstico é realizado por análise genética, na maioria dos casos por cariotipagem. Porém, pode ser realizada hibridização in situ (FISH), análise cromossômica por microarranjo, reação em cadeia da polimerase e sequenciamento genético.
Na suspeita diagnóstica no pré-natal, a análise genética do bebê recém-nascido é obrigatória para confirmação! Na suspeita forte + cariótipo normal, sugere-se a repetição do teste, utilizando um tecido diferente (p.ex. swab bucal).
Alterações cardiovasculares: um problema grave para as pacientes
As alterações cardiovasculares são o problema mais grave associado à Síndrome de Turner, pois aumentam muito o risco de mortalidade. Metade das pacientes apresentam malformações cardiovasculares, e a principal causa de morte de pacientes com a síndrome é a dissecção aórtica.
A hipertensão é comum na vida adulta, ocorrendo em 30-50% dos casos. Outras alterações incluem:
- Anormalidade da valva aórtica – 30% dos casos
- Arco aórtico transversal alongado – 40-50% dos casos
- Coarctação aórtica – 18% dos casos
- Alteração do septo atrial e/ou ventricular – 1-4% dos casos
- Anormalidades venosas sistêmicas – 8-13% dos casos
- Anormalidades venopulmonares – 13-15% dos casos
- Vasculopatia progressiva
- Aumento do intervalo QT – 20-40% dos casos

Prognóstico
Há aumento do risco de diversas condições clínicas, como doenças autoimunes, osteoporose, problemas cardiovasculares graves. Como citamos, as alterações cardíacas estão associadas a um pior prognóstico.
Após avaliar cuidadosamente todas as características clínicas e comorbidades associadas, o tratamento é direcionado para cada condição, visto a ampla gama de fenótipos possíveis. A estimativa de vida é cerca de 10-13 anos a menos na presença da Síndrome de Turner, e o tratamento adequado e precoce melhora a expectativa de vida.
Quer saber mais? Acesse o conteúdo completo da Síndrome de Turner e de outras doenças genéticas no nosso app WeMEDS®. Disponível na versão web ou para download para iOS ou Android.
—
Referências:
Philippe Backeljauw, MD. Clinical manifestations and diagnosis of Turner syndrome. UpToDate®. Sep 2023.
Sociedade Brasileira de Endocrinologia e Metabologia Sociedade Brasileira de Genética Clínica. Síndrome de Turner: Diagnóstico e Tratamento. Junho de 2006.
MAYO CLINIC. Turner syndrome. Disponível em: https://www.mayoclinic.org/diseases-conditions/turner-syndrome/symptoms-causes/syc-20360782. Acessado em: 08/11/2023.









")




{kind=link}