



















Neurofibromatose tipo I
A Neurofibromatose tipo I (NF1) ou doença de von Recklinghausen é uma condição caracterizada por alterações da pigmentação da pele e o crescimento de tumores ao longo dos nervos da pele, cérebro e outras partes do corpo. Os tumores geralmente não são cancerosos (benignos), mas podem causar uma série de sintomas.
A doença ocorre devido a variantes patogênicas no gene NF1. É herdada de forma autossômica dominante – cada filho de um indivíduo com NF1 tem 50% de chance de herdar a variante causadora da doença. Porém, até 50% dos casos diagnosticados são provocados por mutações de novo – aquelas novas, nas quais os progenitores não apresentam a mesma alteração.
A maior parte dos pacientes não apresenta sintomas. Nos sintomáticos, mais de 90% apresentam lesões cutâneas ao nascimento ou durante a infância. O diagnóstico é clínico, e os critérios do NIH de 2021 podem auxiliar.

NIH (National Institute of Health) 2021
O critério diagnóstico de Neurofibromatose tipo I (NIH 2021) é um índice desenvolvido para auxiliar na confirmação diagnóstica da doença em pacientes sob investigação.
Sua elaboração foi proposta com o intuito de revisar os critérios diagnósticos para NF1 de 1987 e 1997, incorporando novas características clínicas e testes genéticos.
Os critérios foram desenvolvidos na National Institute of Health (NIH) Consensus Conference. Foi usado um processo de várias etapas para o desenvolvimento dos critérios clínicos e genéticos mínimos, envolvendo especialistas globais e, posteriormente, envolvendo não especialistas, pacientes e fundações/grupos de defesa de pacientes.
Critérios, Pontuações e Interpretação
O critério diagnóstico de NF1 do NIH (2021) é composto por 7 critérios clínicos. É utilizado em pacientes com suspeita diagnóstica, para diagnosticar ou descartar a doença, e possui alta sensibilidade e especificidade para pacientes acima de 1 ano.
Veja cada um dos critérios:
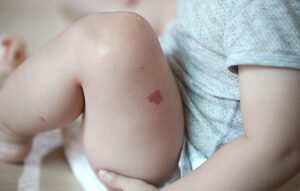
- Seis ou mais máculas café-com-leite com mais de 5 mm de maior diâmetro em indivíduos pré-púberes, e mais de 15 mm de maior diâmetro em indivíduos pós-púberes.
- Sardas na região axilar ou inguinal.
- Dois ou mais neurofibromas de qualquer tipo, ou um neurofibroma plexiforme.
- Glioma da via óptica.
- Dois ou mais nódulos de Lisch da íris identificados por exame com lâmpada de fenda, ou duas ou mais anormalidades coroidais – definidas como nódulos brilhantes e irregulares fotografados por tomografia de coerência óptica (TOC)/refletância próxima ao infravermelho (RPI).
- Uma lesão óssea distinta, como displasia esfenoidal, arqueamento anterolateral da tíbia ou pseudoartrose de um osso longo.
- Uma variante heterozigótica do gene NF1 patogênica, com uma fração do alelo variante de 50% em tecido aparentemente normal, como glóbulos brancos.

A confirmação do diagnóstico de Neurofibromatose tipo I ocorre da seguinte maneira:
– Caso o indivíduo tenha um dos pais diagnosticado com NF1: pontuação em pelo menos 1 critério diagnóstico.
– Caso o indivíduo não tenha um dos pais diagnosticado com NF1: pontuação em pelo menos 2 critérios diagnósticos.
Vantagens vs. Armadilhas do uso dos critérios
Os critérios de NIH 2021 são a versão mais atualizada dos critérios diagnósticos de NF1. Ainda, como vantagem avalia mutações no gene NF1.
Porém, os critérios não apresentam sensibilidade e especificidade satisfatória em pacientes com idade < 1 ano.
Após o diagnóstico, quais os próximos passos?
Os exames complementares são importante para avaliação diagnóstica, avaliação de complicações e exclusão de diagnósticos diferenciais.
Crianças pequenas que apresentam apenas uma manifestação clínica e sem história familiar de NF1 devem continuar a ser monitoradas quanto ao aparecimento de outras manifestações, pois o diagnóstico definitivo geralmente pode ser feito até os quatro anos de idade.
Lembre-se: a confirmação diagnóstica de neurofibromatose tipo I permite o monitoramento e manejo precoce de complicações associadas à doença, minimizando seu impacto.
Para saber mais sobre este e outros critérios diagnósticos, acesse o nosso app WeMEDS®. Disponível na versão web ou para download para iOS ou Android.
—
Referências:
Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. 2021 Aug;23(8):1506-1513. doi: 10.1038/s41436-021-01170-5. Epub 2021 May 19. PMID: 34012067; PMCID: PMC8354850.
Friedman JM. Neurofibromatosis 1. 1998 Oct 2 [Updated 2022 Apr 21]. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1109/
Angelova-Toshkina, D., et al. (2022). Neurofibromatosis type 1: A comparison of the 1997 NIH and the 2021 revised diagnostic criteria in 75 children and adolescents. Genetics in medicine: official journal of the American College of Medical Genetics, 24(9), 1978–1985. https://doi.org/10.1016/j.gim.2022.05.013
















{kind=link}