



















Um estudo publicado dia 27 de abril na revista Nature trouxe atualizações relevantes na Reumatologia. Os pesquisadores identificaram uma alteração genética suficientemente capaz de gerar lúpus eritematoso sistêmico (LES).
Lúpus Eritematoso Sistêmico (LES)
O Lúpus Eritematoso Sistêmico (LES) é uma doença inflamatória autoimune, apresentando-se desde a forma leve até a forma grave. A inflamação ocasionada por essa doença acomete todo organismo do paciente, podendo comprometer diversos órgãos e ou sistemas, como a pele, os rins, coração, pulmões e articulações.
Em um post anterior, detalhamos as características da doença, sinais/sintomas e tratamentos atuais. Recomendamos a leitura (clique aqui). Além disso, no nosso app WeMEDS é possível encontrar informações detalhadas sobre a doença.
Nesse artigo, vamos focar no estudo publicado essa semana.
O LES pode ser monogênico?
O LES é conhecido por sua característica autoimune poligênica. As principais alterações genéticas envolvidas incluem os genes HLA-DR2 e HLA-DR3, deficiência em proteínas da cascata do complemento e receptores de imunoglobulinas e alteração de mecanismos envolvendo citocinas. Porém, estudos recentes apresentam casos de lúpus monogênico, e variantes patogênicas raras fornecem informações importantes sobre os mecanismos da doença.
Um novo estudo publicado na Nature essa semana (27 de abril) mostrou que alterações em TLR7 (toll-like receptor) podem ser causadoras de lúpus. Os receptores toll-like são moléculas transmembrana, presentes em macrófagos, células dendríticas e neutrófilos, importantes para o reconhecimento de patógenos e, consequentemente, para a resposta imune. Para a sinalização intracelular ser ativada, alguns domínios são necessários, como é o caso da proteína MyD88 – presente em todos os TLR.

O estudo: investigação da relação entre TLR e lúpus
Antes de começarmos, um comentário: o artigo é complexo, e cheio de detalhes de genética molecular! Para quem tem interesse nessa área, recomendamos a leitura do artigo completo (aqui). Aqui, tentamos simplificar ao máximo, permitindo o entendimento do conteúdo, sem deixar de lado os achados mais relevantes.
Dito isso, vamos ao estudo.
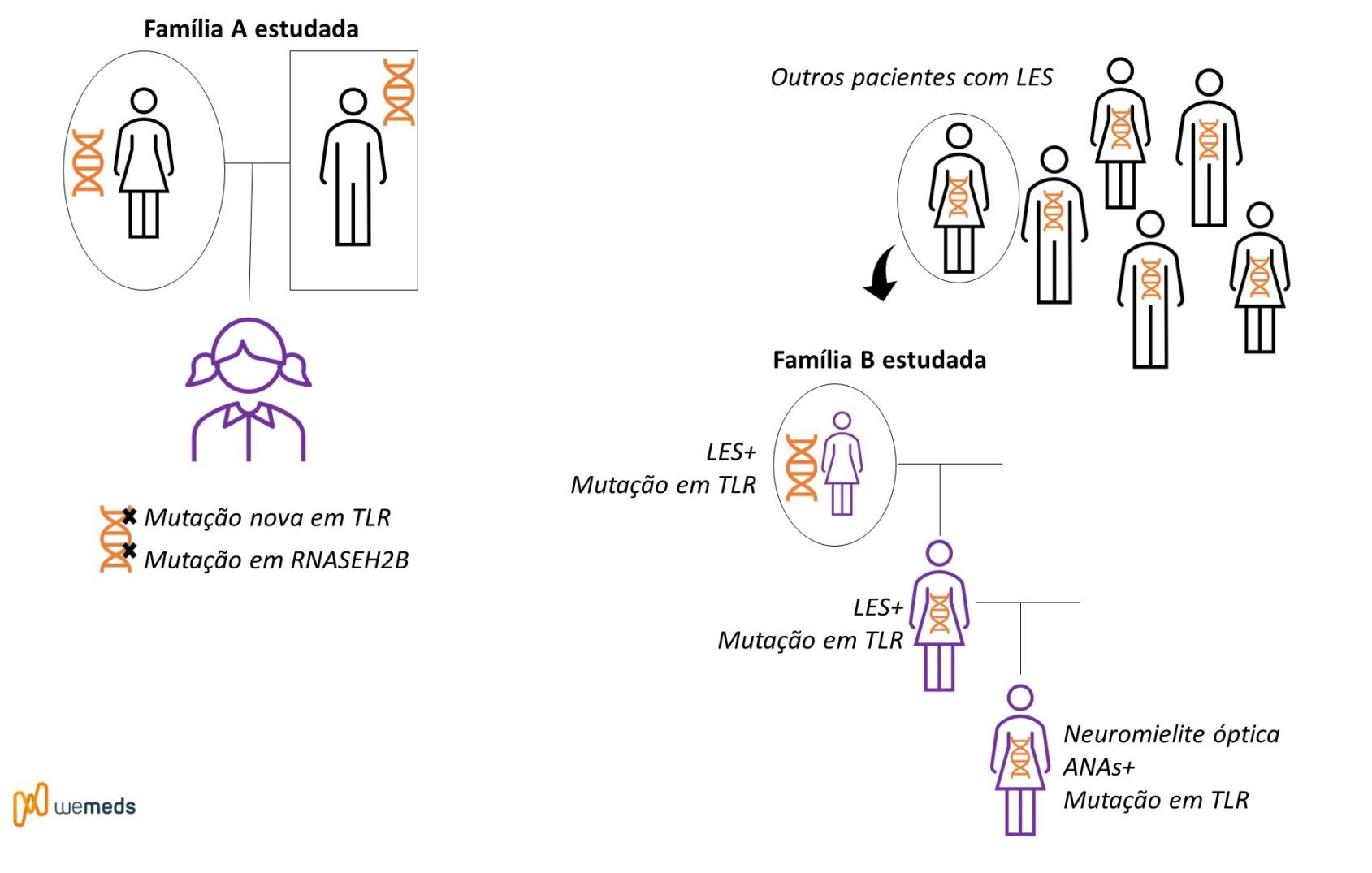
Os pesquisadores realizaram o sequenciamento de todo o genoma de uma menina espanhola que foi diagnosticada com LES aos 7 anos de idade. Apresentava inicialmente trombocitopenia autoimune refratária e níveis elevados de anticorpos antinucleares (ANAs) e hipocomplementemia.
A paciente evoluiu com artralgias inflamatórias, sintomas constitucionais, episódios intermitentes de hemicoreia, insuficiência mitral leve e comprometimento renal após internação com crise hipertensiva.
A análise de bioinformática pós-sequenciamento revelou uma variante genética em TLR7 (nome oficial: p.Tyr264His (Y264H)). Esta variante não estava presente nas bases de dados de variação normal do genoma humano. Os exames adicionais e as análises da família confirmaram que foi uma mutação nova (chamada de novo).
Foram realizadas análises adicionais para variantes raras em 22 genes que podem causar LES humano quando mutados. Essas análises revelaram uma variante no gene RNASEH2B (nome oficial: p.Ala177Thr). Essa variante, quando em homozigose, é conhecida por causar LES. A paciente apresentou essa variante em heterozigose – ou seja, apenas um dos alelos alterado.
Os pesquisadores também realizaram o sequenciamento de outros pacientes com LES, e foram identificadas outras duas variantes em TLR7. Nessa nova família estudada, a mãe do paciente também teve LES a partir de seus vinte e poucos anos e a filha foi diagnosticada com neuromielite óptica (ANAs+). Todos apresentavam a mutação em TLR7 identificada no sequenciamento, mas nenhuma daquelas variantes raras adicionais nos 22 genes causadores do LES foi identificada nessas famílias.
Está ficando um pouco confuso? Veja o esquema abaixo:
Achados encontrados na presença da mutação em TLR
Os autores viram que a presença das mutações em modelos animais foi suficientemente capaz de gerar autoimunidade. Alguns dos achados foram observados em animais com a variante em TLR:
- Trombocitopenia e leucopenia, glomerulonefrite proliferativa e infiltrados linfoides no fígado, glândulas salivares e pâncreas;
- Infiltrados subpleurais, perivasculares e intersticiais nos pulmões;
- Degeneração e necrose de miócitos no músculo panículo da pele;
- Fibrose miocárdica focal;
- Linfomas esplênicos;
- Linfadenite crônica nos gânglios linfáticos e intestino;
- Hiperplasia das placas de Peyer;
- Aumento dos níveis séricos de IFNγ, IL-6, IL-10 e TNF;
- Redução da razão de células T:B;
- Aumento total de células B, plasmócitos e APCs;
- Aumento de células efetoras – CD4+, CXCR3+;
- Aumento da expressão de MHC-II.
Mais informações podem ser encontradas no artigo.
Como a mutação em TLR funciona na patogênese de LES?
Os pesquisadores buscaram identificar se essas variantes patogênicas aumentavam a sinalização de TLR7 e ativação de NF-κB.
Por técnicas de engenharia molecular, os autores viram que as células que tinham um aumento de expressão das mutações apresentavam maior ativação da via de NF-κB em comparação àquelas que não tinham as variantes. Além disso, os autores viram que houve um aumento de afinidade à guanosina, o que reduz o limiar de ativação de TLR.
Ou seja, um achado muito relevante do artigo foi quanto a ativação de TLR.
Os pesquisadores viram que na presença de uma das mutações, o TLR era capaz de estar ativado mesmo na ausência de estimulação. Além disso, a proteína MyD88 (falamos dela no começo do artigo) foi detectada aumentada nos animais com a mutação – o que faz sentido com a ativação de TLR.
Além disso, foi visto que essa sinalização constitutiva fornece um sinal aberrante para as células B autorreativas (que se ligam ao autoantígeno). Em geral, essas células morreriam em cerca de 72 horas, mas a sobrevivência delas é aumentada na presença da variante em TLR.
Quais as conclusões desse trabalho? Devemos procurar mutações causadoras de doença em pacientes com LES?
Os autores destacam a importância de estudos futuros para determinar se os achados são verdadeiros para todos os casos de LES, ou apenas para pacientes nos quais a sinalização excessiva de TLR7 é a via patogênica dominante.
Ainda, as mutações altamente prejudiciais de TLR7 são raras. No entanto, é evidente que o aumento da sinalização de TLR7 é um fator chave em lúpus. Dessa forma, terapias que bloqueiam o próprio TLR7 ou MyD88 podem ser eficazes.
—
Referências:
FERRAZ, E.G. et al. Toll-Like Receptors: regulation of the immune responses. RGO, Rev. gaúch. odontol. (Online) vol.59 no.3 Porto Alegre Jul./Set. 2011.
Brown, G.J., Cañete, P.F., Wang, H. et al. TLR7 gain-of-function genetic variation causes human lupus. Nature (2022). https://doi.org/10.1038/s41586-022-04642-z








: Indicações Terapêuticas e Riscos do Uso Indevido")







{kind=link}